Dystrophin: The Reader’s Digest abridged edition
In spite of a solid grounding in experimental practice and animal models, gene therapy has had a difficult road in the clinic. According to the Journal for Gene Therapy, over 1800 gene therapy-oriented clinical trials have been conducted worldwide since 1989, including 67 Phase III trials – typically the last stage in a drug’s journey to the market. However, only one gene therapy product has been approved for use in patients: Glybera, from Dutch company UniQure, which uses a virus to deliver a replacement for a damaged gene normally responsible for fatty acid metabolism. Importantly, Glybera is only available in the EU, although the company seems confident that they’ll win approval from the FDA in short order.
In other words, pharma and biotech companies are still feeling their way in this new land – but that’s not to say that there aren’t a lot of really cool and promising studies out there. A lot of neat stuff has been bubbling up around muscular dystrophy (MD), which encompasses a family of disorders arising from mutations of the gene encoding the dystrophin protein. Dystrophin is critical to the proper arrangement and stabilization of muscle fibers and, without it, muscle tissue wastes away until patients ultimately perish from failure of the muscles of the heart and/or respiratory system.
Now, something you should know about dystrophin. It’s big – I mean epically big. Burj Khalifa big. Blue whale big. Let me put this in perspective: the median length of a human protein is 375 amino acids, but dystrophin contains a whopping 3,677. The DNA encompassed by the gene spans 0.07% of the total human genome, and the process of transcribing the gene into a protein-coding messenger RNA molecule takes SIXTEEN HOURS. This is a damn long gene – the kind that would make Marcel Proust or David Foster Wallace proud.

It’s just slightly smaller than this picture, in other words.
But here’s the cool part – you don’t need the whole thing.
The dystrophin RNA needs to undergo a process known as ‘splicing’ before it gets translated into protein, which entails the clipping and stitching together of 79 protein-coding segments known as ‘exons’. In some MD cases, a whole exon or multiple exons might go missing, but even a tiny insertion or deletion of nucleotides in one of these exons can have catastrophic consequences for how the rest of the protein gets made or even halt protein production entirely. But scientists have learned that you can make short strings of chemically-modified ‘antisense’ nucleic acids that will actually bind directly to those damaged or misplaced exons, causing the splicing machinery to ignore them and just move along to the next exon.
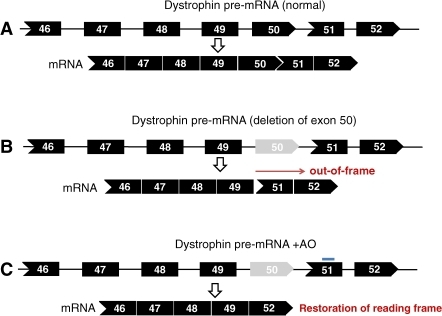
This turns out to not be as problematic as one might think – leaving out a tiny chunk of a behemoth like dystrophin is, of course, a hell of a lot less damaging than gumming up the whole protein in mid-sequence – and the early trials have given some cause for optimism. For example, Sarepta Therapeutics now has a Phase II clinical trial underway in the US to test the efficacy of their drug eteplirsen, which knocks out defects in exon 51 – one of the more common causative mutations for Duchenne muscular dystrophy (DMD), the most common form of MD. GlaxoSmithKline is gunning for the same target with an antisense molecule of their own, GSK2402968, which is currently making its way through both Phase II and Phase III clinical trials.
Both drugs have been initially associated with small but statistically significant performance improvements with regard to muscle function in DMD patients, and it seems like other companies are keen to queue up in developing gene therapies that operate along similar lines. And given that there are a broad range of DMD mutations, and that each exon will most likely require its own targeted exon-skipping antisense molecule, there should be plenty of room to operate.
Unless, of course, you say “The hell with this noise – I’m just gonna make myself a tiny, pocket-sized dystrophin that I can cram into a virus and force patients to express that instead!” That sounds nice, because then you don’t really have to worry about targeting each individual exon in each individual patient. It also sounds like total crazy talk. Especially given that the standard gene therapy viruses can only hold enough genetic material to encode a protein on the order of 1500 or so amino acids.
But many biologists just happen to be that kind of crazy, and not without cause – one patient has been identified who was naturally missing nearly half of her dystrophin gene, and yet she exhibited only relatively mild symptoms and these at a late age. Since then, scientists have been banging away at the idea of creating a viral delivery-friendly, stumpy ‘microdystrophin’ gene that remains sufficiently functional to combat muscle-wasting in DMD patients. Unfortunately for us dog lovers, dogs happen to be the best available ‘large mammal’ model for characterizing the performance of these types of gene therapies. And sadly, many of the sacrifices of these pioneers were made in vain – early microdystrophin constructs were successfully delivered, and even gave rise to truncated protein in treated animals, but with no particular improvements in muscle function… in some cases, muscle performance even suffered as a result of treatment.
A new study in Molecular Therapy gives a bit more cause for hope, however. Dongsheng Duan’s team at the University of Missouri School of Medicine synthesized viruses containing a profoundly abridged version of dystrophin – a mere 30% of the original sequence – and used it to treat six dogs with muscular dystrophy. In each animal, they only dosed one muscle so that they could compare directly against the untreated counterpart in the same dog.
Within two months, they could specifically detect microdystrophin being produced in every single treated muscle. More importantly, microdystrophin proved capable of interacting with the appropriate protein partners in muscle tissue, and seemed to repair many of the abnormalities normally observed in dystrophic tissues.
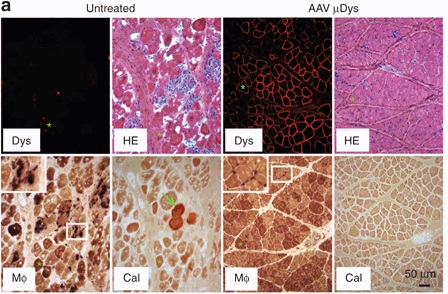
As you can see, a lot of the disorganization and other mishegas (e.g. calcification and immune cell infiltration) that’s going on in the dystrophic tissue (left panels) has been addressed in the microdystrophin-treated muscle (right panels). On the other hand, the physical benefits were less readily apparent. There were no clear improvements in the force of muscle contraction, and although the treated muscle performed significantly better than the untreated in tests of repeated contraction, it was still far from acceptable levels for normal performance.
Nevertheless, this seems like a big step towards demonstrating the principle that a virus-friendly microdystrophin might become a viable option for the broader treatment of DMD patients. It’s abundantly clear that the current minigene is unready for clinical development and that scientists can now look forward to a protracted process of optimization (a.k.a. trial and error), but the authors also want to emphasize the primary positive takeaway point: “Our results have cleared uncertainty on microdystrophin therapy [that have] arisen from other dog studies.”
Ladies and gentlemen – start your editing!